
© Columbia University, all rights reserved.

Quantitative Proteomics and Metabolomics Center



Innovations
Shotgun proteomics is a strategy with broad applicability using liquid chromatography-mass spectrometry (LC/MS). These robust and flexible platforms allow the design or large analyses in clinical patient samples, cell culture or animal model projects without the stringent restrictions on experimental design imposed by isotopic labeling.
Methodology
In a typical experiment, proteins are extracted and digested with proteolytic enzymes. Following reduction, alkylation of cysteines, digestion of peptides is performed. Peptides are separated on a Vanquish Neo liquid chromatograph (LC) coupled to an Orbitrap Q Exactive HF or Exploris 240 mass spectrometer. This system has been demonstrated to exhibit exceptional performance by other groups and in our own published work (see publications tab).
Shotgun proteomics is a strategy with broad applicability using liquid chromatography-mass spectrometry (LC/MS). These robust and flexible platforms allow the design or large analyses in clinical patient samples, cell culture or animal model projects without the stringent restrictions on experimental design imposed by isotopic labeling.
Methodology
In a typical experiment, proteins are extracted and digested with proteolytic enzymes. Following reduction, alkylation of cysteines, digestion of peptides is performed. Peptides are separated on a Vanquish Neo liquid chromatograph (LC) coupled to an Orbitrap Q Exactive HF or Exploris 240 mass spectrometer. This system has been demonstrated to exhibit exceptional performance by other groups and in our own published work (see publications tab).
Background in Quantitative Proteomic Analysis
Early approaches to quantitative proteomics used a comparison of 2-dimensional electrophoretic gels (2D gels) coupled with mass spectrometry, then progressed to the use of isotopic labeling (e. g. ICAT, iTRAQ, SILAC) in a shotgun liquid chromatography/mass spectrometry (LC-MS and LC-MSMS) strategy. All of these approaches have some limitations. The gel-based approaches tend to be cumbersome to use, place restrictions on experimental design and address a limited subset of proteins. The isotopic methods are expensive, place stringent limits on experimental design, the type of samples that may be analyzed, and often rely on weak isotopic signatures. Consequently, there is increasing interest in using so-called "label-free" methods to make proteome-wide comparisons. This technique is an ideal discovery tool for understanding biological processes, stem cells, developing biomarkers for a variety of uses, comparing affinity purifications and immunoprecipitations and studying biofluids such as urine, saliva, plasma, serum and cerebrospinal fluid.
Early approaches to quantitative proteomics used a comparison of 2-dimensional electrophoretic gels (2D gels) coupled with mass spectrometry, then progressed to the use of isotopic labeling (e. g. ICAT, iTRAQ, SILAC) in a shotgun liquid chromatography/mass spectrometry (LC-MS and LC-MSMS) strategy. All of these approaches have some limitations. The gel-based approaches tend to be cumbersome to use, place restrictions on experimental design and address a limited subset of proteins. The isotopic methods are expensive, place stringent limits on experimental design, the type of samples that may be analyzed, and often rely on weak isotopic signatures. Consequently, there is increasing interest in using so-called "label-free" methods to make proteome-wide comparisons. This technique is an ideal discovery tool for understanding biological processes, stem cells, developing biomarkers for a variety of uses, comparing affinity purifications and immunoprecipitations and studying biofluids such as urine, saliva, plasma, serum and cerebrospinal fluid.
Posttranslational Modifications
Posttranslational modifications (PTMs) such as phosphorylation are critical to our understanding of the biology of cells and tissues. Large scale profiling of PTM patterns is necessary to achieve a systems biology understating of pathways and networks.
Phosphorylation
Phosphorylation of amino acid residues in proteins especially serine, threonine and tyrosine is a driver of signal transduction in cells and typically an overarching regulator of protein function. The modulation of protein phosphorylation states by networks of kinases and phosphatases leads to rapid changes in patterns of phosphorylation. Very often regulatory phosphorylation sites have a substoichiometric level of site occupancy, increasing the difficulty of detecting such sites by mass spectrometry. The approach currently used in proteomics is to enrich for phosphorylated peptides using chemical means in digests of proteins from cells, tissues and biological fluids. The understanding of phospho-signaling networks is critical to our understanding of fundamental cellular processes and disease pathology.
Posttranslational modifications (PTMs) such as phosphorylation are critical to our understanding of the biology of cells and tissues. Large scale profiling of PTM patterns is necessary to achieve a systems biology understating of pathways and networks.
Phosphorylation
Phosphorylation of amino acid residues in proteins especially serine, threonine and tyrosine is a driver of signal transduction in cells and typically an overarching regulator of protein function. The modulation of protein phosphorylation states by networks of kinases and phosphatases leads to rapid changes in patterns of phosphorylation. Very often regulatory phosphorylation sites have a substoichiometric level of site occupancy, increasing the difficulty of detecting such sites by mass spectrometry. The approach currently used in proteomics is to enrich for phosphorylated peptides using chemical means in digests of proteins from cells, tissues and biological fluids. The understanding of phospho-signaling networks is critical to our understanding of fundamental cellular processes and disease pathology.
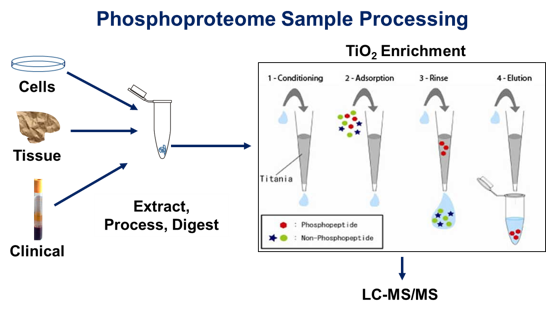
Typical enrichment and analytical
strategy for studies of the phosphoproteome of cells, tissues and
biological fluids as applied in laboratory, translational and clinical
applications
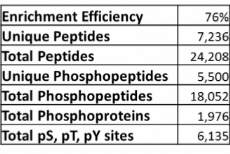
Example data from phosphopeptide enrichment of a proteins lysate from a hepatocellular carcinoma cell line.
Targeted Analyses with Parallel Reaction Monitoring (PRM)
We can use the PRM techniques to select a list of ions arising from peptides from proteolytic digests selected in the quadrupole region of the Exploris 240 mass spectrometer and transferred to the HCD cell of the instrument where fragmentation takes place. Mass/charge ratio is measured in the orbitrap mass analyzer. This provides full MS/MS spectra of the targeted peptides with high resolution and accuracy, providing the ability to perform quantitation often using isotopically-labeled peptide standards.
PRM helps to eliminates interferences from other ions, provides high accuracy and attomole-level limits of detection and quantification and can be applied to panels of particular proteins in targeted studies for laboratory, translational and clinical applications.
We can use the PRM techniques to select a list of ions arising from peptides from proteolytic digests selected in the quadrupole region of the Exploris 240 mass spectrometer and transferred to the HCD cell of the instrument where fragmentation takes place. Mass/charge ratio is measured in the orbitrap mass analyzer. This provides full MS/MS spectra of the targeted peptides with high resolution and accuracy, providing the ability to perform quantitation often using isotopically-labeled peptide standards.
PRM helps to eliminates interferences from other ions, provides high accuracy and attomole-level limits of detection and quantification and can be applied to panels of particular proteins in targeted studies for laboratory, translational and clinical applications.