
© Columbia University, all rights reserved.

Quantitative Proteomics and Metabolomics Center

The proteome is the
expressed protein complement of a cell, matrix, organelle, tissue,
organ, or organism. It includes all isoforms and posttranslational
variants and varies with time. The overall technical approach in
proteomics was enabled by two major technical advances: the ability to
sequence genomes and the ability to analyze proteins by mass
spectrometry. Quantitative proteomics defines
the differences in expression of proteins among different biological
states (e.g., control vs. treatment, healthy vs. disease, specific
genotype vs. wild type) or for affinity purifications. We have
established this center to apply these emerging technologies to a wide
range of biomedical research studies. We use label-free shotgun
profiling with liquid chromatography/mass spectrometry (LC-MS/MS) to
perform these comparisons.
The focus of the center is the identification of
proteins and metabolites with differential quantitative expression in
cells, tissues or in protein affinity purifications, and we have applied
this technique employing mass spectrometry to many different research
problems with a strong focus on clinical,
translational applications and basic science. Particular emphasis is on
the quantitative analysis of posttranslational modifications such as
phosphorylation.



A wide variety of proteomes can be processed
including cells, tissues, organelles, biofluids (e.g. plasma,
cerebrospinal fluid, urine), and affinity pull-downs. Bacterial, yeast,
insect and mammalian proteomes have been effectively studied. With
nanogram amounts of material we can create a complex profile of
quantitative relative expression for almost any biological system.
Accurate mass and retention time data allows mining of large datasets
for unique biological insights based on large-scale protein profiling.
We do projects with groups at
Columbia University and at other institutions across the nation. All
internal and external research groups are treated equally. We
welcome collaborations equally with academic and industry partners
leading to new funding and scientific opportunities.
Background - Quantitative Discovery Proteomics by Mass Spectrometry
Practical Systems Biology Approach to Quantitative Proteomics Widely Applicable



Call us at 646-558-0007
While we follow many approaches,
for proteomics and biomarker development we emphasize large-scale
profiling technologies that offer -
< Flexible experimental design with no limits on numbers of treatments and replicates - providing high statistical power
< No need for the expense of costly isotopically labeled amino acids that increase the economic risk in sample preparation
< Simple experimental protocols in which the chemistry does not restrict the experimental design of the biology component of the experiment
< Easy application to patient samples or affinity pull-downs
< Flexible experimental design with no limits on numbers of treatments and replicates - providing high statistical power
< No need for the expense of costly isotopically labeled amino acids that increase the economic risk in sample preparation
< Simple experimental protocols in which the chemistry does not restrict the experimental design of the biology component of the experiment
< Easy application to patient samples or affinity pull-downs

Contact Center Director, Lewis M. Brown to discuss a collaborative project or a grant proposal under development.
Contact by email: LB2425@columbia.edu

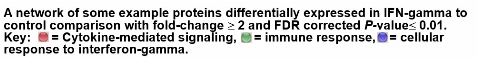
See Wobma, et al. 2018. Biomaterials, 167, 226-234.
Also, please see our metabolomics page for information about that technology.


Our Sponsors
Department of Biological Sciences, Columbia University
Dean of Science, Columbia University
Executive Vice President for Arts and Sciences, Columbia University
Executive Vice President for Research, Columbia University
Department of Biomedical Engineering, Columbia University
The Fu Foundation School Engineering & Applied Science, Columbia University
Columbia Stem Cell Initiative
Department of Medicine, Columbia University
Our External Funding Sources
New York State Stem Cell Science Board (NYSTEM)
National Institutes of Health
National Science Foundation
Department of Defense
Collaborating Institutes
Columbia University Chemical Probe Synthesis Facility
Data Science Institute, Columbia University
Department of Biological Sciences, Columbia University
Dean of Science, Columbia University
Executive Vice President for Arts and Sciences, Columbia University
Executive Vice President for Research, Columbia University
Department of Biomedical Engineering, Columbia University
The Fu Foundation School Engineering & Applied Science, Columbia University
Columbia Stem Cell Initiative
Department of Medicine, Columbia University
Our External Funding Sources
New York State Stem Cell Science Board (NYSTEM)
National Institutes of Health
National Science Foundation
Department of Defense
Collaborating Institutes
Columbia University Chemical Probe Synthesis Facility
Data Science Institute, Columbia University